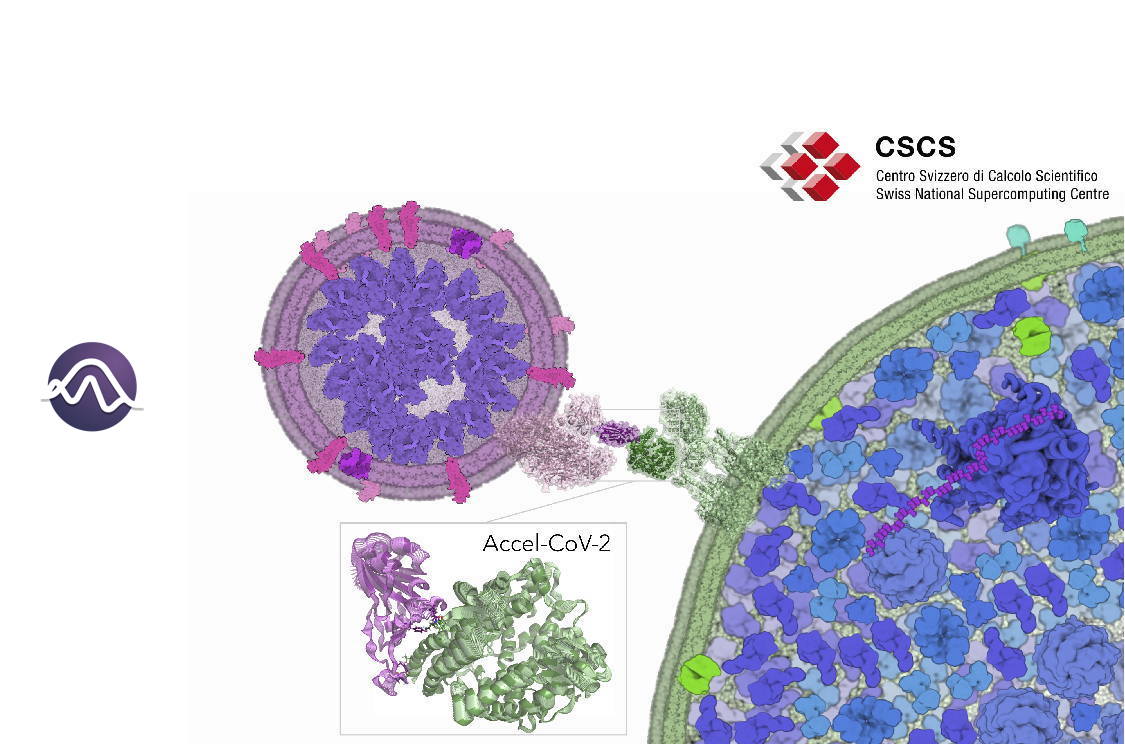
A team of researchers from IQCC has been awarded computational time at the Piz Daint supercomputer of the ETH Zurich/CSCS (Switzerland) to explore the molecular and dynamical SARS-CoV-2/hACE2 recognition and inhibition mechanisms. In the framework of the Accel-CoV-2 project, the team will perform extensive accelerated Molecular Dynamics (aMD) simulations to access millisecond motions that can be key to elucidate the essential molecular details of how the spike protein of SARS-CoV-2 recognizes and dynamically interacts with the host cell receptors (human angiotensin-converting enzyme 2 (hACE2)). The insights gained from this aMD simulations can provide key atomistic details for designing new therapeutic agents for COVID-19 treatment.
The spike protein (S) of SARS-CoV-2 plays a key role in recognizing and facilitating the virus entry into human host cells. This protein is composed of different domains that adopt a variety of conformations relevant for the mechanism of infection. The receptor-binding domain (RBD) of SARS-CoV-2 is the responsible of specifically recognizing human angiotensin-converting enzyme 2 (hACE2). The intrinsic dynamic character of proteins present a strong influence on biomolecular recognition mechanisms. Unraveling the conformational dynamics of proteins and biomolecular assemblies is key to gain insight into biomolecular and pharmacological mechanisms. However, conformational changes occurring in a protein prior and upon association with other (bio)molecules are diverse and not obvious to predict when little information is available. The process of SARS-CoV-2/hACE2 recognition involves a series of complex conformational changes that take place in a variety of timescales (from ns to ms and beyond). Being able to access millisecond time scales motions may be key to identify key steps of the SARS-CoV-2 infection mechanism and also reveal cryptic druggable pockets in both SARS-CoV-2 and hACE2. Therefore, elucidating the essential molecular details of how SARS-CoV-2 recognizes and dynamically interacts with the host, will provide relevant information on the mechanisms of virus infection. This knowledge can be applied for designing, developing or repurposing therapeutic agents that target the spike protein/hACE2 interaction to prevent SARS-CoV-2 infection.
Molecular Dynamics (MD) simulations are used to characterize the biomolecular conformational dynamics and identify hidden conformations with cryptic druggable pockets. The size and complexity of conformational changes of SARS-CoV-2 spike protein S/hACE2 complex hampers the ability of MD to efficiently explore the conformational landscape of this biomolecular assembly in a reasonable computational time. Enhanced sampling techniques speed up MD simulations. In particular, accelerated MD (aMD) provides unconstrained enhanced sampling to efficiently explore the conformational space of biomolecules. Hundreds of nanoseconds of aMD are able to capture millisecond time scale events without a priori definition of a reaction coordinate. aMD is perfectly suited for shedding light on the unexplored conformational space of RBD/hACE2 in a reasonable computational time.
Goals of the project: The team of researchers from IQCC aims to use aMD simulations to explore and map the conformational landscape of spike protein/RBD in complex with hACE2 to elucidate the essential molecular details of how SARS-CoV-2 recognizes and dynamically interacts with the host receptor, gaining insight into inhibition mechanisms. To this end, extensive aMD simulations will be run at the GPUs of Piz Daint CSCS supercomputer. aMD simulations will provide access to millisecond timescale motions that can reveal elusive spike protein S/hACE2 conformations and interactions. These simulations will be analyzed to discover druggable hidden conformations and cryptic pockets in both RBD and hACE2 proteins and identify key protein-protein interactions that can be used to accelerate the design of therapeutic agents that target spike protein S/hACE2 interaction to counteract SARS-CoV-2 infection.
The research team that conceived the idea is composed by Jordi Soler-Parpal, Carla Calvó-Tusell, Miguel A. María-Solano, Marc Garcia-Borràs, and Ferran Feixas. This project combines knowledge from different areas (Computational Biochemistry, Biotechnology, and Physics). The team combines expertise in metalloenzymes, computational chemistry, homology modelling, molecular biology, and accelerated molecular dynamics. The project has been positively evaluated and granted with computational resources at the Swiss National Supercomputing Centre (CSCS). The accomplishment of the project goals will be possible thanks to the acceleration provided by GPUs of Piz Daint CSCS supercomputer. The advances of the project will be periodically reported. The team will be in contact with the Scientific communication committee of the IQCC and Càtedra de Cultura Científica i Comunicació Digital for disseminating the project results. All data will be made publicly available through online repositories.
Girona, April 30, 2020
More info: ferran.feixas@udg.edu