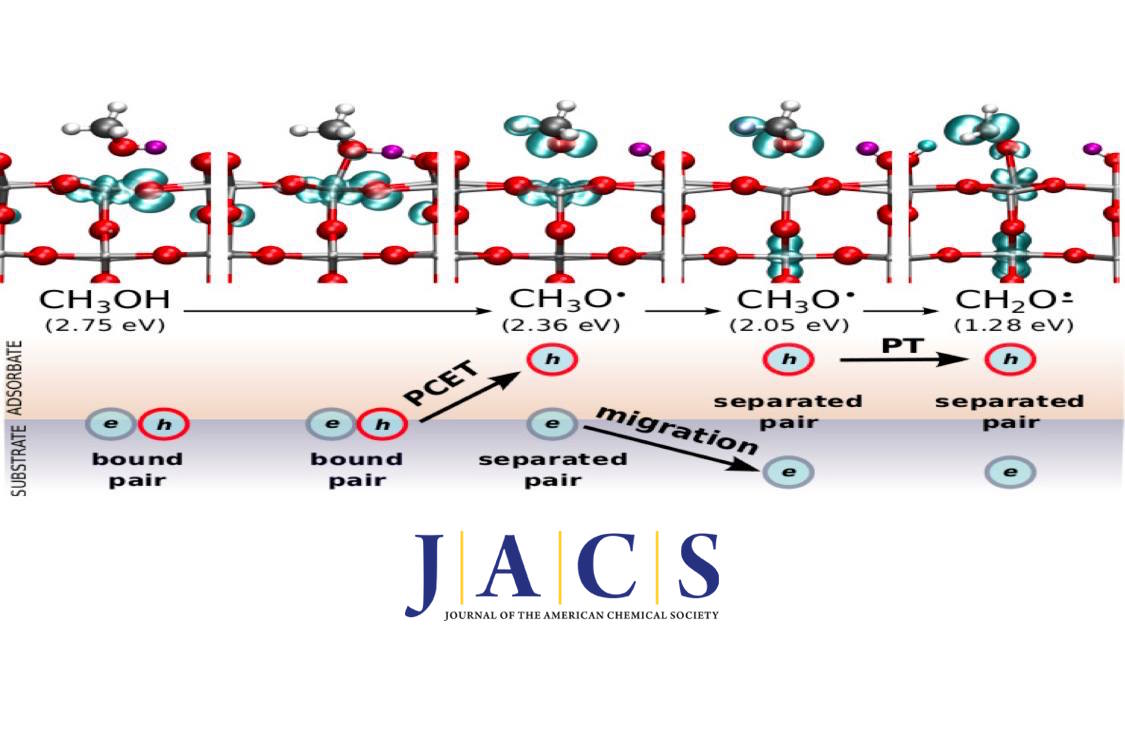
CH3OH on a single-crystal rutile TiO2(110) surface is a widely studied model system for heterogeneous photocatalysis. The oxidation to CH2O is stepwise and involves a CH3O intermediate. These photocatalytic processes are usually modeled assuming that the hole-electron (h-e) pairs generated during the excitation are independent, ie they separate before the reactive photocatalytic step. Here we show that bound excitons are crucial in the first step of the photocatalytic oxidation. The first O–H dissociation step follows an excitonic interfacial proton-coupled electron transfer mechanism where the photogenerated exciton is bound, and the h is transferred to the adsorbate. The O–H dissociation paths associated with other h–e pairs are unreactive, and the moderate experimental efficiency is due to the different reactivity of the h–e pairs. The excited-state CH3O intermediate further deactivates through a seam of intersection between the ground and excited states. It can follow three different paths, regeneration of adsorbed CH3OH or formation of the ground-state CH3O anion or an adsorbed CH2O radical anion. The third channel corresponds to photochemical CH2O formation from CH3OH, where a single photon induces one electron oxidation and transfer of two protons. These results expand the current view on the photocatalysis of CH3OH on TiO2(110) by highlighting the role of excitons and showing that adsorbed CH3OH may also be an active species in the photocatalytic oxidation to CH2O.
The results were published today in Journal of the American Chemical Society:
A. Migani and L. Blancafort
“Excitonic Interfacial Proton-Coupled Electron Transfer Mechanism in the Photocatalytic Oxidation of Methanol to Formaldehyde on TiO2 (110)”
J. Am. Chem. Soc. 2016, ASAP-
[abstract]
DOI: 10.1021/jacs.6b11067