
Dual-Function Enzyme Catalysis for Enantioselective Carbon–Nitrogen Bond Formation
This study reports a new biocatalytic platform for creating new carbon-nitrogen bonds in a very selective and efficient way, with applications in the pharmaceutical and industrial fields. The selective formation of these bonds is relevant for the synthesis of new bioactive and pharmacological compounds. To achieve this, a new enzyme has been engineered to catalyze these chemical transformations under mild conditions. This project is a collaboration between a research group of the California Institute of Technology: Dr. Zhen Liu, Andrew Z. Zhou, Dr. Kai Chen and Professor Frances H. Arnold and members of the Institute of Computational Chemistry and Catalysis of the University of Girona, Carla Calvó-Tusell and Dr. Marc Garcia-Borràs.
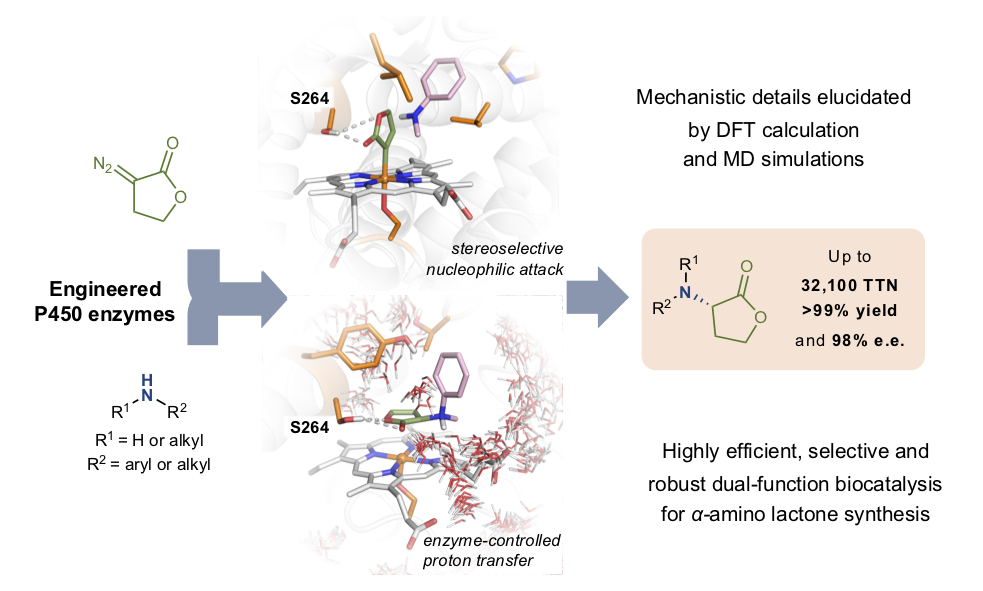
Asymmetric carbene N–H insertion is a powerful method for the preparation of chiral amines, however, it has traditionally shown limited enantioselectivity. Haem proteins can perform carbene N–H insertion, but the asymmetric protonation in an active site replete with proton sources is the most challenging step.
In this work, it was demonstrated that engineering cytochrome P450 enzymes can catalyze the abiological C–N bond forming reaction with excellent activity and selectivity (up to 32,100 total turnovers, >99% yield and 98% e.e.) to prepare a series of bioactive ?-amino lactones. The engineered P411 (P450 with the axial cysteine ligand coordinated to iron is replaced by serine) enzymes acted as dual-function biocatalysts that promoted the transfer of the lactone carbene to amines and exerted excellent stereocontrol in the subsequent protonation step, in a single active site.
The researchers from the IQCC performed computational modelling to understand how this new system works. Specifically, extensive molecular dynamics (MD) simulations and density functional theory (DFT) calculations in selected P411 variants were performed to gain insight into the mechanism of this enzymatic transformation. Computational studies uncovered the detailed mechanism of this process, explaining the critical role of the serine residue at position 264 for achieving high activity and selectivity. In particular, MD simulations showed that S264 established a persistent H-bond with the lactone ester group that fixes the orientation of the lactone carbene in the active site. Therefore, the engineered active site controls the conformation of the lactone carbene, yielding to an enantiospecific N-nucleophilic attack for the ylide formation. Once dissociated from the iron, the ylide intermediate can rapidly react with active site water molecules to achieve a stereoselective proton rearrangement in the enzyme active site prior to product release.
The study was recently reported in Nature Chemistry:
Z. Liu, C. Calvó-Tusell, A.Z. Zhou, K. Chen, M. Garcia-Borràs, and F.H. Arnold
“Dual-function enzyme catalysis for enantioselective carbon–nitrogen bond formation”
Nat. Chem. 2021, 13, 1166-1172
DOI: 10.1038/s41557-021-00794-z
The work was highlighted by the Nature Chemistry journal cover, in its December 2021 issue. The cover was designed by Carla Calvó-Tusell and illustrates the laboratory enzyme evolution, represented in different layers, to finally achieve an engineered P450 enzyme that works as a dual-function biocatalyst with a single active site.

Girona, December 3, 2021
For more information: gestor.iqcc@gmail.com