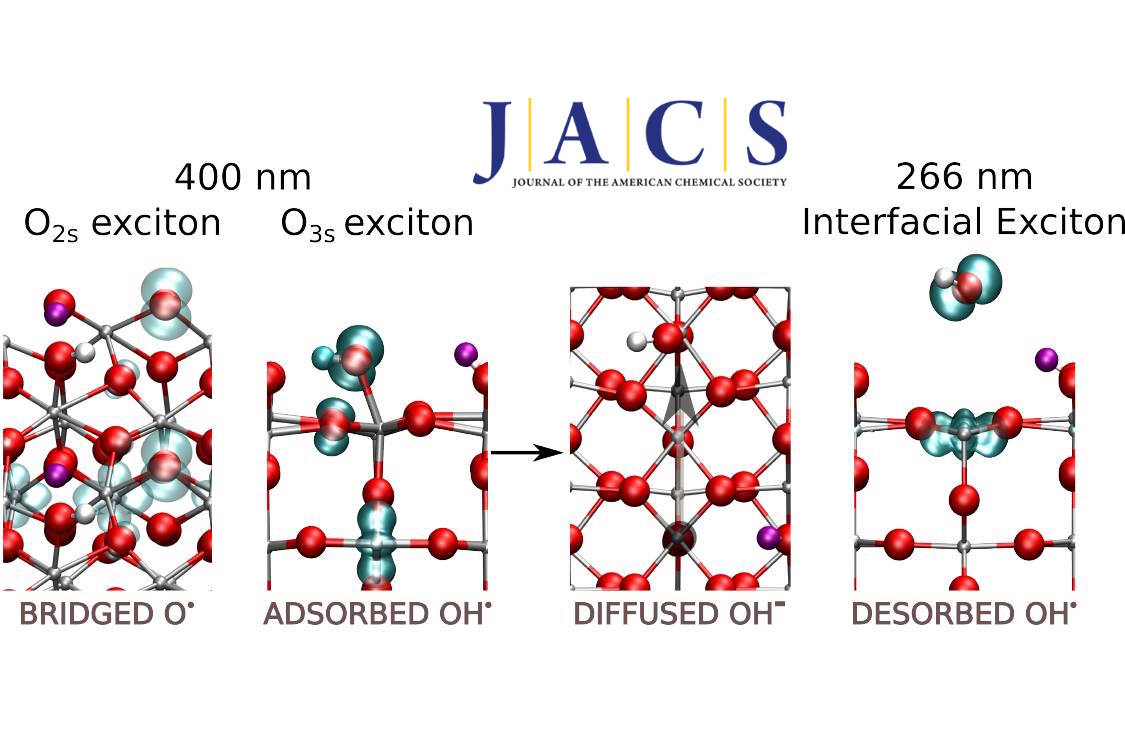
The photocatalytic O–H dissociation of water absorbed on a rutile TiO2(110) surface in ultrahigh vacuum (UHV) is studied with spin-polarized density functional theory and a hybrid exchange-correlation functional (HSE06), treating the excited-state species as excitons with triplet multiplicity. This system is a model for the photocatalytic oxidation of water by TiO2 in an aqueous medium, which is relevant for the oxygen evolution reaction and photodegradation of organic pollutants. We provide a comprehensive mechanistic picture where the most representative paths correspond to excitonic configurations with the hole located on three- and two-coordinate surface oxygen atoms (O3s and O2s). Our picture explains the formation of the species observed experimentally. At near band gap excitation, the O3s path leads to the generation of hydroxyl anions which diffuse on the surface, without net oxidation. In contrast, free hydroxyl radicals are formed at supra band gap excitation (e.g., 266 nm) from an interfacial exciton that undergoes O–H dissociation. The oxidation efficiency is low because the path associated with the O2s exciton, which is the most favored one thermodynamically, is unreactive because of a high propensity for charge recombination. Our results are also relevant to understand the reactivity in the liquid phase. We assign the photoluminescence measured for atomically flat TiO2(110) surfaces in an aqueous medium to the O3s exciton, in line with the proposal based on experiments, and we have identified a species derived from the O2s exciton with an activated O2s–Ti bond that may be relevant in photocatalytic applications in an aqueous medium.
The results were published today in Journal of the American Chemical Society (http://www.dx.doi.org/10.1021/jacs.7b05121):
A. Migani, and L. Blancafort
“What Controls Photocatalytic Water Oxidation on Rutile TiO2 (110) under Ultra-High-Vacuum Conditions?”
J. Am. Chem. Soc. 2017, 139, 11845-11856 [abstract]
DOI: 10.1021/jacs.7b05121