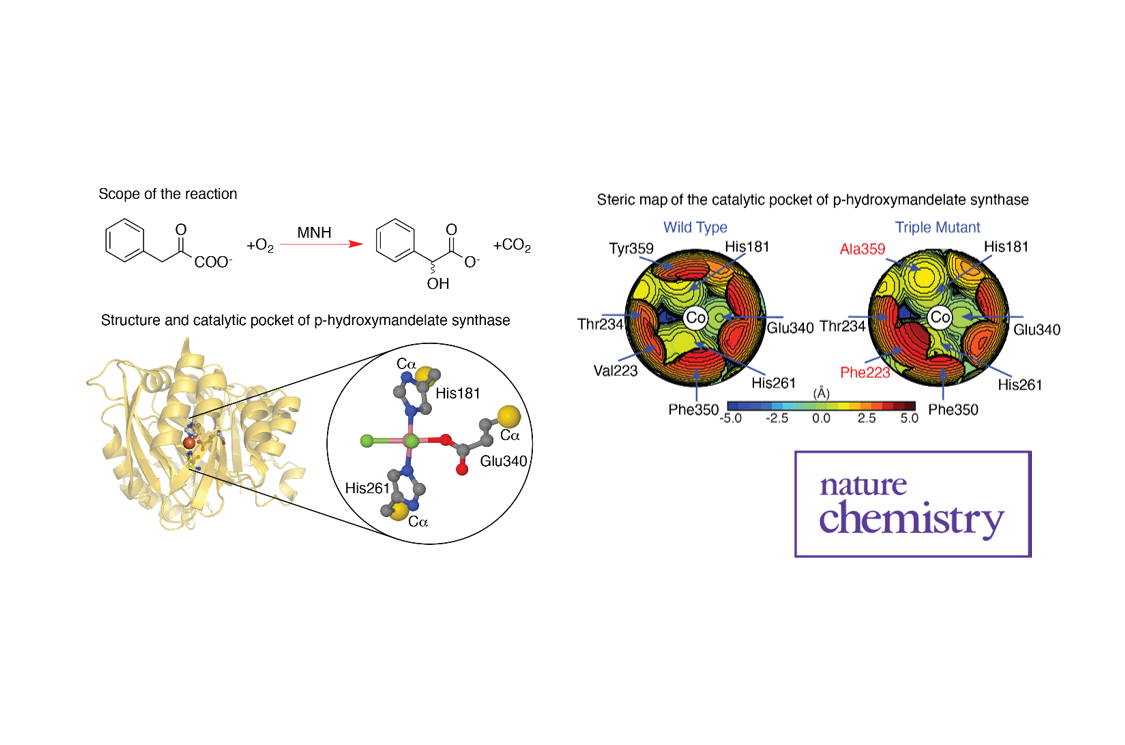
Making chemical reactions faster or more selective can be achieved by catalysts, which can be biomolecular enzymes or transition-metal molecules. Engineering these catalysts to provide specific functionality on demand has been a long-term dream by many chemists. In a recent contribution by a team of researchers from Girona, Italy and Saudi Arabia, this dream has been brought one step closer to become reality. The team focused on finding numerical descriptors that correlate (bio)molecular structure with reactivity, which allowed them to create topographic steric maps that provide a three-dimensional image of the catalytic pocket (that area of the catalyst where the actual reaction takes place). This online tool is available to the wider chemical community to quickly explore structural modifications, especially with the help of quantum-chemical calculations based on density functional theory, to rationalize the behaviour of known catalysts and/or to design improved catalysts.
The IQCC researcher involved, Albert Poater, adds: “We have been working more than ten years on this project, and it is very satisfying to see the work finally out there”. The study was published recently in Nature Chemistry:
L. Falivene, Z. Cao, A. Petta, L. Serra, A. Poater, R. Oliva, V. Scarano, and L. Cavallo
“Towards the online computer-aided design of catalytic pockets”
Nature Chem. 2019, online, ASAP [abstract]
DOI: 10.1038/s41557-019-0319-5