
On May 9, 2019, the Universitat de Gironaconferred the Honorary Doctorate degree (Doctor Honoris Causa) of the university to Prof. Evert Jan Baerends (VU University Amsterdam) and biologist Prof. Sovan Lek (University Toulouse III). Prof. Baerends is of course well known in the field of chemistry, in particular for his contributions to density functional theory and chemical bonding analyses. He is also the main author of the Amsterdam Density Functional (ADF) program, and has contributed greatly to the careers of many (current and previous) IQCC members, among which Prof. Miquel Solà, Prof. Marcel Swart, and Prof. Jordi Poater. The ceremony was streamed live and can be viewed here. Below you can find the laudatio and Prof. Baerends’ speech.
Scientific merits
To summarize in few words the scientific career of Professor Baerends is a very complex task, since his main contribution, beyond the tangible achievements, lies in his capacity to transmit to his collaborators his enthusiasm for the discovery and his perennial commitment to rigor and truth. Evert Jan Baerends has put his life career at the service of Science as a way of acquiring knowledge and also as an instrument of its diffusion to the new generations, and he has done it with passion, as a simple extension of his passion for life in all its facets.
Evert Jan Baerends is an emeritus professor of the Vrije Universiteit Amsterdam. He is one of the most prominent researchers in the field of theoretical and computational chemistry nowadays. He was born in 1945 in Voorst, Friesland, a region of The Netherlands with its own language like Catalonia. He did his PhD in the Vrije Universiteit Amsterdam under the supervision of Prof. Pieter Ros. During his PhD he had to deal with transition-metal complexes. He started to study computationally these type of complexes with the traditional Hartree-Fock method, but he rapidly realized that this method was not providing reliable results. At this time, physicists studied solid crystals containing metals using the X? method, a method based on the density functional theory (DFT). So, he decided to investigate whether methods based on the DFT could be used in transition metal chemistry. It was a brave, somewhat quixotic (for a man who physically reminds the Quijote), decision since nobody used the DFT method in chemistry before, and, consequently, he had to program his own code to perform the simulations. Pragmatic and efficient numerical approaches were implemented by Baerends to generate his own Hartree-Fock-Slater (HFS) program, which later became the Amsterdam Molecule (AMOL) code, and currently is used widely by computational chemists as the Amsterdam Density Functional (ADF) program. The program stands out by unique features such as Slater orbitals and the pioneering use of precise numerical integration, density fitting, linear scaling, and parallelization techniques. Since the 70s, Prof. Baerends and his collaborators, with a special mention to the late Prof. Tom Ziegler, have been demonstrating the utility of DFT tools for computational chemistry studies. Twenty years after the first DFT calculations by Baerends, the Gaussian program incorporated the DFT methods. It was in the Gaussian 92 release of this program. Thanks to the Gaussian implementation, DFT methods became highly popular in the theoretical and computational chemists’ community. Today DFT has become the computational quantum mechanical modelling method most used in physics, chemistry, and material science to investigate the electronic structure of atoms, molecules, and condensed phases. In 1998 the Nobel prize of Chemistry was awarded to Walter Kohn for “for his development of the density-functional theory” and to John A. Pople “for his development of computational methods in quantum chemistry.” DFT reached this level of popularity thanks to the work of Prof. Baerends and collaborators who proved the utility of this method. Many of us consider that it would have been fair to have included Prof. Baerends in the list of awardees of the 1998 Nobel prize of Chemistry. The ADF program was later complemented by the BAND DFT program which extends the molecular treatment to periodic systems. Also in this field Prof. Baerends made significant contributions.
We will briefly refer to other relevant works of Evert Jan Baerends. He made a substantial contribution to elucidate the connection between properties of the exchange–correlation (xc) potential and that of the so-called exchange–correlation hole. In this way, he was able to improve the calculation of response properties and time-dependent DFT. He also explored the relation between the density matrix and the xc-hole. The density matrix functionals, which are the basis of the density-matrix functional theory, were developed further in later studies opening a promising avenue to improve current DFT methods. Analysis of the chemical bonding and chemical interactions, a central aspect of chemistry, was carried out by him using Morokuma’s energy decomposition analysis as implemented in ADF. The treatment of relativistic e?ects on electronic structure was also mastered by Prof. Baerends. He developed the e?cient and widely used Zeroth Order Regular Approximation (ZORA) which is very well-suited for application with density functional theory. Finally, the Baerends group developed also a multilevel approach entirely based on DFT. He particularly contributed to the field of property calculations with the frozen-density embedding (FDE) for molecules in solvents for properties like UV-Vis spectra, electron paramagnetic resonance hyperfine coupling constants, and circular dichroism spectra.
All these huge scientific contributions have been gathered in more than 450 papers that have received more than 50000 citations. His h index is 98. Among many distinctions, let us mention that he received in 2010 the prestigious Schrödinger Medal of the World Association for Theoretically Oriented Chemists (WATOC) for this pioneering contributions to the development of computational density functional methods and his fundamental contributions to density functional theory and density matrix theory.

Speech by Prof. Baerends
Esteemed Rector Magnificus,
President of the Board of Trustees of the University of Girona,
Esteemed Authorities,
Professors, Students and Members of the University Staff,
Ladies and Gentlemen,
It is a great honor for me to receive the doctorate honoris causa from your university. For several reasons.
One of the reasons is the special place that Catalunya has held for me since a long time. I think contacts started in the nineties when Dr. Miquel Sola, now professor Miquel Sola, spent time as a postdoc in Amsterdam. I spent three summer periods as an Iberdrola Fellow at the Universitat Autonoma de Barcelona, being hosted by professors Joan Bertran, Mariona Sodupe and Vicenç Branchadell, renewing also contacts with the Theoretical and Computational Chemistry group of the University of Girona at that time. The contacts have not been limited toUniversitat Autonoma de Barcelonaor Girona, I remember memorable visits to Tarragona and to the University of Barcelona. I cannot record all contacts, but I should mention that since that time a strong connection with the Netherlands has built up over the years, with many fruitful exchanges.
Another reason to enjoy this award, apart from the personal honor it brings to me, is the tribute it constitutes to the field of science I am involved with, and am representing today, namely chemistry. It is a truism that science in general, and chemistry and physics in particular, have shaped our modern world. Maybe I should include biology, and then in its wake also medical science, but let me note a bit chauvinistically that biochemistry has hugely impacted biology and medicine. It is not only the technical achievements of the natural sciences that have impacted our lives. Their influence is much broader and deeper. They shape our culture and our outlook on life. They also are a source of beauty. Chemistry offers the beauty of chemical structures. I just have to mention the DNA molecule, with its intricate double helix structure. And chemistry is a creative art. It creates many new beautiful structures, never before realized in nature.
On this occasion, however, I want to stress the lesson science teaches us about our mental outlook. Given that this award is in chemistry, in particular in theoretical chemistry, and then in the subfield of density functional theory, I think it is appropriate to highlight the role of open mindedness, the lack of prejudice, the flexibility of mind to adopt new paradigms, even when not yet completely established. In science, any preconception runs the risk of being thoroughly demolished. It appears that all obvious conceptions in whatever field one is interested in, are bound to be dispelled by further investigation.
By way of example, before I focus on theoretical chemistry, let me remind you of a very well known but actually very esoteric problem, that the most famous scientist ever has set himself. This most famous scientist is Einstein, you will agree. The question he struggled with is: can anything go faster than light? Isn’t that an irritating question at a time the highest speeds were achieved by trains running ca. 100 km per hour, compared to the speed of light of 300 000 km per second? I am afraid a grant application by Einstein would not have been rated high at “usefulness for society”.
Now we all know that Einsteins results have revealed staggering insights in such fundamental issues as the nature of space and time. But also position determination with GPS needs Einsteins theory of relativity. The utterly useful and very widely employed GPS devices that serve as the everyday navigation equipment in our automobiles rely on the theory of relativity! GPS has also revolutionized navigation at sea. Sextants and celestial navigation are not even taught any more at maritime schools! Often, the more fundamental insights are, the more revolutionary useful they prove to be. The lesson is: never reject a notion or question off-hand, however irritating or esoteric it may seem, be open minded and unprejudiced.

Let me focus then on chemistry, and to make a long story short, on my own field of the theory of chemical bonding and structure. Let me start with the picture of bonding by electron pairs as developed by Gilbert Lewis (Figure 1), which I think every high-school student will remember from his chemistry classes. In the first decades of the twentieth century Lewis pictured chemical bonds as arising from electron pairs (Figure 2), and the remaining electrons pairing up in so-called lone pairs.

This model rationalized a large number of known facts. In the first place the predominance of compounds with an even number of electrons. In the second place the explanation within this model of the valences of the elements when combined with the rule that an atom tries to collect 8 electrons (the octet rule). But of course there is also something ridiculous in this theory: why would negatively charged electrons, which according to electrostatics will repel each other, get together to form a bond? Moreover, just at the time Lewis was developing these ideas, the Bohr theory of the hydrogen atom, which pictures electrons as fast moving particles circling the atomic nucleus, like planets are circling the sun, was astonishingly successful in predicting quantitatively the spectra of the H atom. In a sense Bohr’s model marks the beginning of the efforts that would lead, about a decade later, to quantum mechanics. Quantum mechanics is one of the two great theories of physics to emerge in the twentieth century, and from the physics side there was only ridicule and contempt for Lewis’s model. And not only from physics. Also many physical chemists felt that Lewis’s electron pair theory was too simplistic, the basis of electronic structure theory should rest firmly in the quantum theoretical treatment of the motions of the electrons. This may have cost Lewis the Nobel prize, he is often referred to as the most famous and deserving chemist who never got the Prize. But we should recognize that Lewis has had enormous impact in chemistry. His concepts were so useful and such an excellent basis for theorizing about bonding in molecules that even today every chemist knows what Lewis structures are. Much of his thinking, for instance his generalization of the concepts of acidity and basicity, to donation and acceptance of electron pairs, are now fully vindicated by quantum chemistry. Lewis is another example that we should beware of hasty condemnation and be open minded and unprejudiced.
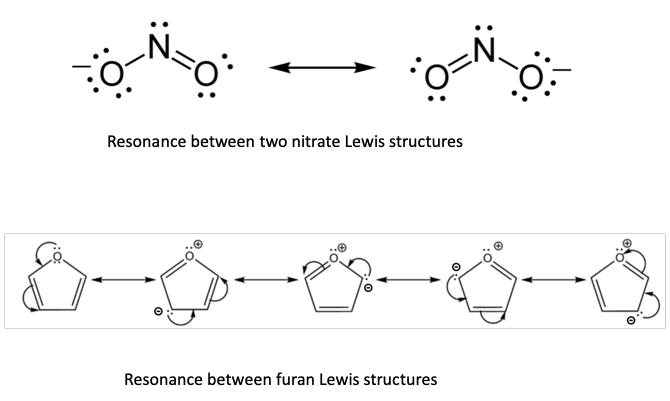
It has been Linus Pauling (Figure 3) who has done an admirable job of tying in quantum mechanics with Lewis’s ideas, even before computers were powerful enough to actually perform quantum mechanical calculations on the motions of the electrons. He realized that even without computation one can give chemists a feeling for the relative importance of quantum mechanical effects by considering what he called “resonance” among various electronic structures. These contributions, also called valence bond structures, translate into simple pictures the quantum mechanical superposition principle (Figure 4). Many chemists who never studied quantum mechanics in any depth, and would think of it as a rather esoteric physical theory, were as a matter of fact practicing quantum mechanical concepts in their daily thinking and talking about the structures and reactivity of their compounds. I am ashamed to admit that as a young theoretical chemist I have made the error of ridiculing Lewis’s and Pauling’s models with their “unphysical” electron pairs, and the arrows indicating how these pairs might hop around in order to create other contributing resonance structures. So modesty is something that has to be acquired. We should be open minded and modest enough to keep in mind that what seems at first sight to be absurd and contrary to accepted wisdom may eventually turn out to contain a lot of truth.
Lewis and Pauling created a paper and pencil method for judging bonding and structures of compounds. But when computers became sufficiently powerful, it became possible to actually carry out the complicated and time-consuming calculations that are necessary to solve the quantum mechanical equations with sufficient accuracy. Sufficient accuracy was denoted “chemical accuracy”, something like 0.1 kcal/mol in the energy. That would make it possible to actually do “chemistry on the computer” or “in silico”. This development started slowly in the fifties and then made great leaps in the subsequent decades, mostly through the astonishing improvements in computer technology. Expectations (in my opinion unrealistic) were very high that eventually, maybe even soon, reducing chemistry to a computational science might become a reality. There was also a clear paradigm: calculations should be done “ab initio”, that is by solving the equations purely by mathematical and numerical methods, without empirical parameters. We may use the picture of Thomas Kuhn, who distinguished periods of “normal science” and “scientific revolutions”. In our case then, the paradigm was given, no scientific revolution would be needed. We could envisage a period of “normal science” where theoretical chemists would work hard on the given task of developing increasingly sophisticated and efficient techniques of solving the given equations. They would eventually reach the holy grail of “chemical accuracy”.
As Kuhn describes, scientists usually enjoy such normal science. The goal is clear, it is easy to see who makes the smartest contributions to solving the set problem. All efforts can be directed towards developing methods to solve the given problem. No need to waste time and energy on debates about the underlying science or the goals to be reached. However, the downside is that the scientists no longer are so modest as to recognize that maybe the chosen path is not the right one, or at least not the only one. They may hate colleagues who will doubt the accepted paradigm, with a professional and sometimes even personal hatred.
This is what happened when in the seventies and eighties a new paradigm was provided in our science by the advent of density functional theory. We give it the acronym DFT. This is not the place to go into any detail about this theory. Let me just note that it provided a great simplification of the equations to be solved. The big drawback was the absence of a simple straightforward path to the exact solutions. In theory DFT did afford exact solutions. But often “in theory” is almost a euphemism for “not in practice”. That is also to some extent the case with DFT, but not completely. It has not been made exact, by no means, but it is sufficiently accurate to be eminently useful. My career has entirely revolved around this particular approach. DFT has become thoroughly successful, by now maybe 90% of all quantum chemical calculations are DFT calculations. But the initial reception of DFT was utterly hostile. How did a small group of theoretical chemists dare to challenge the path forward that had been chosen by the very large majority of theoretical chemists, namelyab initiocalculations? If I look back on my career, I like to divide it into two halves: the first twenty years (seventies and eighties) I was met, as a DFT practitioner and developer, with scorn and contempt. I remember a conference, organized by two well known professors in our field, in the mid eighties, where I was invited as a speaker. But they were harassing me so much at that meeting, that afterwards their students invited me to dinner, telling me how embarassed they were by this treatment. That is an illustration of the danger that old and established scientists are in. They may get too much entrenched in their pet theories and methods and may start to consider anything else as an aberration. It is also a beautiful illustration that, fortunately, young people tend to be much more open minded. That I am receiving this doctorate honoris causa today I consider a great honor for me personally, but at the same time I think it is recognition of the importance of the scientific revolution that has led to the predominance of DFT in our field, in particular for large systems.
But what I want to single out most is that again we see demonstrated that as scientists, we should never be rigid and prejudiced. We should be rigorous in our proofs and deductions, but that is a different matter. Things may always turn out to be just the opposite of what we think. Actually, they very often do. The ab initioapproach looked so admirably clear and rigorous, while DFT was, and still is, far from straightforward and indeed somewhat muddled. Many people do not fully understand it, and there is still much to be criticized. But it has provided an enormous boost to the application of quantum chemical methods in chemistry. When I started my scientific career, most of the chemistry professors at my university, and surely at other universities as well, would question any “chemical relevance” of the theoretical chemists, with their penchant for mathematics and physics and computer work. That attitude amongst experimental colleagues has totally disappeared. They now enjoy to have computational input to their work. That is almost hundred percent owing to DFT.
So DFT is my last and telling example of the great virtue and even necessity in science to be open minded and without prejudice. It gives me great joy and satisfaction that this award underlines how this principle can lead, as so often in history, to unexpected and important progress.
Thank you for your attention.
Evert Jan Baerends,
Girona, May 9, 2019