Pedro currently leads a research team at IQCC mainly working on the development and implementation of new tools for wavefunction analysis, with the aim of connecting classical chemical concepts with modern ab initio methods.
Back in 2001 we published a decomposition method of the Hartree-Fock energy (HF) in mono and diatomic contributions in the framework of the Quantum theory of Atoms in Molecules (QTAIM), later known as Interacting Quantum Atoms. The difficulty of the associated numerical integrations motivated the exploration of other alternative definitions for the atom in the molecule based on so-called fuzzy atoms. In 2004 we showed how the classical formulation of bond order or overlap populations could be obtained in a general way in the general framework of the fuzzy atoms. In later works this methodology extended to the decomposition of both HF (2005) and Kohn-Sham DFT (2007) molecular energies.
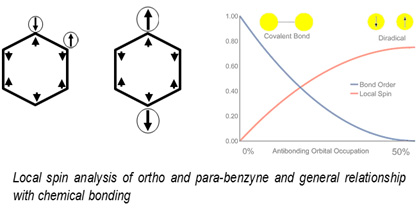
During the last years we have shown how to obtain other local physical magnitudes of interest through analysis of the wavefunction, such as local hardness (2007-08) or local spin (2012-14), the latter being used as a quantification of polyradical character (2014).
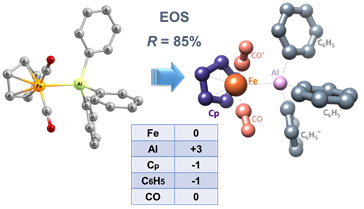
The implementation of Mayer’s effective atomic orbitals for a general wavefunction (2009, 2013) has allowed us to address the recurrent problem of obtaining oxidation states (2015, 2018) from first principles. We have thus developed the so-called effective oxidation states (EOS) analysis.
We have also proposed new atomic definitions less arbitrary in the fuzzy atoms framework (2013) as well as a significant number of applications, most recent ones involving transition metal systems with intricate bonding situations. Some more recent works have dealt with the application of these analysis techniques for the development of new correlation indicators (2014, 2016) oriented to the design of new DFT and DMFT functionals.
We are also currently joining forces with the group of Josep M. Luis focusing on the design and implementation of novel energy-based origin-independent decomposition of linear and nonlinear optical properties
All the new methodology that we’ve developed during the last 15 years is implemented in our own code, APOST-3D, which we have made available to the scientific community.








